Cystic Fibrosis: New Therapy Profoundly Improves Quality of Life
A new therapy could profoundly improve cystic fibrosis patients’ quality of life, say doctors.
Patients suffering from cystic fibrosis often die before their 40s as mucus clogs and damages their lungs and leaves them prone to infection.
A major international trial on 1,108 patients, in the New England Journal of Medicine, showed a combination of drugs could bypass the genetic errors that cause the disease and may increase life expectancy.
Errors in sufferers’ DNA – inherited from their parents – damage the microscopic machinery that controls salt and water levels in the linings of the lungs.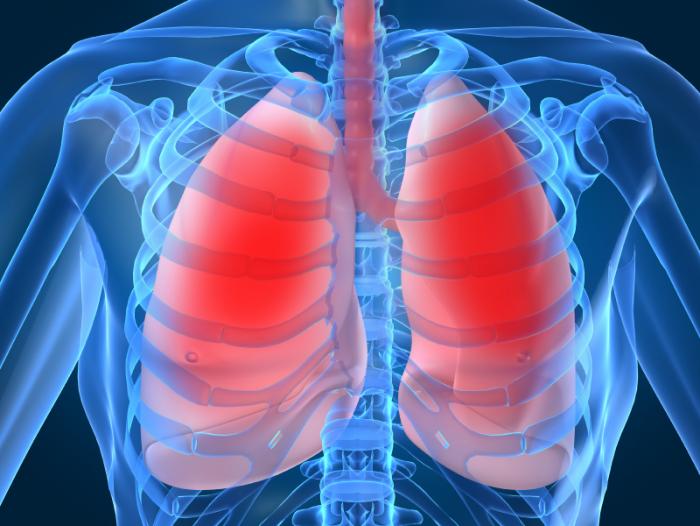
The result is a thick mucus that inexorably damages the lungs.
Antibiotics help prevent infection and drugs can loosen the mucus, but nothing deals with the fundamental problem for most patients.
The combination of drugs – lumacaftor and ivacaftor – were designed to repair that microscopic machinery.
The trial showed that those patients given the cocktail for 24 weeks had better lung function.
Cystic fibrosis also affects the mucus lining in the gut so the doctors were pleased to see the patients also gained weight in the trial.
There are however, many types of error in the DNA that can culminate in cystic fibrosis.
This treatment combination should work on around half of patients, while one of the drugs on its own corrects a small proportion of errors.
New treatments are still required for the remaining patients.
The therapy is being examined by regulators around the world.